Hydridy přechodných kovů
Hydridy přechodných kovů jsou sloučeniny obsahující atomy vodíku vázané na přechodné kovy. Hydridy vytváří většina přechodných kovů a některé takové sloučeniny mají využití jako katalyzátory, Označení „hydrid“ zde má širší význam: některé jsou kyselé (například hydrid tetrakarbonylu železa, H2Fe(CO)4), zatímco jiné mají hydridovou povahu, kdy obsahují ionty H− (například hydrid zinečnatý, ZnH2).
Rozdělení
Binární hydridy
Řada přechodných kovů tvoří sloučeniny s vodíkem, nazývané binární hydridy, jejichž molekuly obsahují pouze kov a hydridový anion. Tyto sloučeniny bývají polymerní a nerozpustné ve všech rozpouštědlech. Často jsou elektricky vodivé, podobně jako kovy. Mnohdy jde o nestechiometrické sloučeniny. Elektropozitivní (Ti, Zr, Hf, Zn) a některé další kovy vytváří hydridy se stechiometrickými vzorci MH nebo MH2 (M = Ti, Zr, Hf, V, Zn). Nejlépe prozkoumané jsou binární hydridy palladia, snadno vytvářející monohydrid. Plynný vodík může difundovat skrz Pd za tvorby PdH jako meziproduktu.[1]
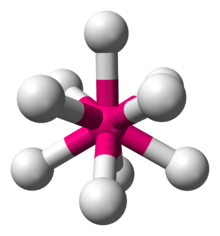
9 v soli K2ReH9.[2]
Ternární hydridy
Ternární hydridy mají obecný vzorec AxMHn, kde A+ je kation alkalického kovu nebo kovu alkalických zemin, například K+ nebo Mg2+. Příkladem může být nonahydridorhenistan draselný (K2ReH9), sůl obsahující dva ionty K+ a anion ReH 2−
9 . Další příklady jsou anionty sloučenin Mg2FeH6 a Mg2NiH4. Některé z těchto aniontových polyhydridů vyhovují pravidlu 18 elektronů a jiné nikoliv. Vzhledem ke své vysoké mřížkové energii obvykle nejsou rozpustné v žádném rozpouštědle, výjimku představuje K2ReH9.[3]
Koordinační sloučeniny
Nejběžnějším druhem hydridů přechodných kovů jsou komplexní sloučeniny obsahující kromě hydridů i další ligandy; spektrum možných přídavných ligandů je široké. Takovéto sloučeniny mohou vytvářet téměř všechny kovy, ovšem stříbro, zlato, kadmium a rtuť vytváří jen málo komplexů s přímými vazbami M-H; tyto hydridy navíc bývají nestálé. K průmyslově využívaným hydridům patří HCo(CO)4 a HRh(CO)(PPh3)3, sloužící jako katalyzátory hydroformylací.

HFeCl(dppe)2 je jedním z nejlépe získatelných hydridů přechodných kovů.

První molekulové hydridy přechodných kovů popsal ve 30. letech 20. století Walter Hieber se svými spolupracovníky, šlo o H2Fe(CO)4 a HCo(CO)4. V 50. letech byly třemi skupinami popsány další takové hydridy: HRe(C5H5)2 (Geoffrey Wilkinson), HMo(C5H5)(CO)3 (Ernst Otto Fischer) a HPtCl(PEt3)2 (Joseph Chatt).[4] Nyní jsou známy tisíce takových sloučenin.
Shlukové hydridy
Podobně jako hydridokomplexy má mnoho shluků koncové (spojené jednou vazbou M–H) hydridové ligandy. Hydridy mohou rovněž tvoři můstek mezi dvěma kovy, například u [HW2(CO)10]−. Dekakarbonyldihydridotriosmium, H2Os3(CO)10, má koncové i dvojitěmůstkové hydridové ligandy. Hydridy také mohou spojovat trojúhelníkovité části shluků, jak je tomu v [Ag3{(PPh2)2CH2}3(μ3-H)(μ3-Cl)]BF4.[5]
[Co6H(CO)15]− obsahuje „intersticiální“ hydrid, nacházející se uprostřed Co6 osmistěnu. Určení struktury shlukových hydridů může být obtížné, což se ukázalo u Strykerova činidla, [Cu6(PPh3)6H6].[6]
Příprava
Přenos hydridu
Nukleofilní hydridy prvků hlavní skupiny přeměňují halogenidy a kationty přechodných kovů na odpovídající hydridy:
- MLnX + LiBHEt3 → HMLn + BEt3 + LiX
Tyto přeměny probíhají jako podvojné záměny a hydricita produktu je zpravidla nižší než u donoru hydridu. K běžným donorům patří borohydrid sodný a hydrid lithnohlinitý. V laboratořích se často používají „smíšené hydridy“, jako například triethylborohydrid lithný a hydrid bis(2-methoxyethoxy)hlintolithný. Hydridy alkalických kovů, jako je hydrid sodný, obvykle nejsou vhodné.
Eliminační reakce
Hydridy zprostředkovávají beta-hydridové a alfa-hydridové eliminace. První z těchto reakcí často ukončuje homogenní polymerizace. Tímto způsobem lze také připravit některé komplexy hydridů přechodných kovů z organolithných a Grignardových činidel:
- MLnX + LiC4H9 → C4H9MLn + LiX
- C4H9MLn → HMLn + H2C=CHC2H5
Oxidační adice
Oxidační adice divodíku na přechodné kovy v nízkých oxidačních číslech jsou součástmi mnoha hydrogenací, například u Vaskova komplexu:[7]
- IrICl(CO)(PPh3)2 + H2 ⇌ H2IrIIICl(CO)(PPh3)2
Oxidační adice také mohou probíhat u dikovových komplexů:
- Co2(CO)8 + H2 ⇌ 2 HCo(CO)4
Oxidačních adicí se může účastnit mnoho různých kyselin, což ukazuje například adice HCl na Vaskův komplex:
- IrICl(CO)(PPh3)2 + HCl → HIrIIICl2(CO)(PPh3)2
Heterolytické štěpení divodíku
Některé hydridy kovů vznikají reakcemi komplexů kovů s vodíkem za přítomnosti zásad. Při těchto reakcích se nemění oxidační čísla kovů; lze je považovat za štěpení H2 na hydrid (který se váže na kov) a proton (jež se naváže na zásadu).
- MLnx+ + zásada + H2 ⇌ HMLn(x−1)+ + Hzásada+
Meziprodukty těchto reakcí jsou pravděpodobně komplexy divodíku. Bifunkční katalyzátory takto aktivují H2.
Termodynamické vlastnosti
| Komplex | Disociační energie (kJ/mol) | pKa |
|---|---|---|
| H-CpCr(CO)3 | 257 | 13,3 |
| H-CpMo(CO)3 | 290 | 13,9 |
| H-CpW(CO)3 | 303 | 16,1 |
| H-Mn(CO)5 | 285 | 14,1 |
| H-Re(CO)5 | 313 | 21,1 |
| H-FeH(CO)4 | 283 | 11,4 |
| H-CpFe(CO)2 | 239 | 19,4 |
| H-CpRu(CO)2 | 272 | 20,2 |
| H-Co(CO)4 | 278 | 8,3 |
Při nahrazení CO fosfinovými ligandy se disociační energie sníží o přibližně 6 kJ/mol.
Vazby M-H mohou uvolňovat protony, vodíkové radikály, nebo hydridy.[9]
- HMLn ⇌ MLn− + H+
- HMLn ⇌ MLn + H
- HMLn ⇌ MLn+ + H−
Hydrid kovu může být slabou kyselinou i slabým donorem H−, případně silný v jedné z těchto oblastí, ovšem ne v obou. H− síla hydridu, také nazývaná jako hydricita, souvisí se sílou hydridu jako Lewisovy zásady; ne všechny hydridy jsou silné Lewisovy zásady. Zásaditost hydridů může být velmi odlišná, podobně jako pKa protonů. Hydricitu lze měřit pomocí heterolytického štěpení vazeb kovu se zásadou o známé pKb a měřením vytvořené rovnováhy; hydrid přitom nesmí heterolyticky nebo homolyticky reagovat se svými molekulami za uvolnění vodíku; komplex by homolyticky reagoval se sebou, pokud by homolytické štěpení vazeb M-H bond bylo méně výhodné než homolytické štěpení vazeb H-H. I v případech, kdy je odolnost vazeb vůči takovým reakcím dostatečná, mohou komplexy reagovat radikálově.
- 2 HMLnz ⇌ 2 MLnz + H2
Komplex bude reagovat heterolyticky, pokud je zároveň silnou kyselinou a silným hydridem. Dojde přitom k disproporcionaci za vzniku dvojice komplexů s oxidačními stavy lišícími se o dva elektrony. Následně může docházet k dalším elektrochemickým reakcím.
- 2HMLnz ⇌ MLnz+1 + MLnz−1 + H2
Některé komplexy za přítomnosti zásad heterolyticky štěpí vodík. Některé z nich vytvářejí produkty dostatečně kyselé na to, aby mohly být deprotonovány. Výchozí komplex může být dvouelektronově redukován vodíkem a zásadou. I v případech, kdy hydrid není dostatečně kyselý, aby se dal deprotonovat, se může homolyticky štěpit podobně jako u výše uvedené jednoelektronové redukce.
2 deprotonace: MLnz + H2 + 2 zásady ⇌ MLnz−2 + 2 H+zásada
Deprotonace následovaná homolýzou: 2MLnz + H2 + 2 zásada ⇌ 2 MLnz−1 + 2H+zásada
Hydricita
Afinita hydridového ligandu k Lewisově kyselině se nazývá hydricita:
- MLnHn− ⇌ MLn(n+1)− + H−
Protože se hydridy v roztocích nevyskytují jako stabilní anionty, tak se tato rovnovážná konstanta (a s ní související volná energie) musí počítat z měřitelných rovnováh. Výchozím bodem je hydricita protonu, která má v acetonitrilovém roztoku hodnotu −318 kJ mol−1:[10]
- H+ + H− ⇌ H2 ΔG298 = −318 J mol−1
Většina kationtů má k H− menší afinitu než proton; jako příklady lze uvést:
- [Ni(dppe)2]2+ + H− ⇌ [HNi(dppe)2]+ ΔG298 = −264 kJ mol−1
- [Ni(dmpe)2]2+ + H− ⇌ [HNi(dmpe)2]+ ΔG298 = −212 kJ mol−1
- [Pt(dppe)2]2+ + H− ⇌ [HPt(dppe)2]+ ΔG298 = −222 kJ mol−1
- [Pt(dmpe)2]2+ + H− ⇌ [HPt(dmpe)2]+ ΔG298 = −178 kcal mol−1
Tyto hodnoty ukazují, že [HPt(dmpe)2]+ by měl být silným donorem hydridů, jelikož [Pt(dmpe)2]2+ je poměrně stálý.[11]
Kinetika a mechanismus
Rychlosti přenosu protonů na komplexy a mezi nimi jsou často nízké.[12]
Mnoho hydridů nelze zkoumat Bordwellovými cykly, k určení jejich termodynamických vlastností se tak používají kinetiky příslušných reakcí. Hydridy přechodných kovů první řady většinou mívají nejrychlejší kinetiky, které se v dalších periodách zpomalují.
Struktura
Určení struktury kovových hydridů může být obtížné, protože hydridové ligandy nevykazují silné interakce s rentgenovým zářením, na rozdíl od kovů. Vzdálenosti M-H byly z tohoto důvodu, obzvláště v prvních studiích, často podhodnocené. Přítomnosti hydridů se běžně vyvozovaly z nepřítomností ligandů na koordinačních místech. Struktury hydridů kovů byly zpravidla zkoumány neutronovou difrakcí. protože vodík velmi dobře rozptyluje neutrony.[13]
Je známo mnoho komplexů s koncovými hydridy, ale i vícejaderné sloučeniny, které obvykle obsahují můstkové hydridy. Řada těchto komplexů můstkových hydridů vytváří oligomery, příklady mohou být Strykerovo činidlo,[14] [(Ph3P)CuH]6 a shluky, jako je [Rh6(PR3)6H12]2+.[15] Na konci vazebné struktury se nachází molekuly divodíku. Prvním případem dobře popsaného neklasického komplexu tohoto typu byl [W(PR3)2(CO)3(H2)].[16][17]
Rentgenová difrakce obvykle neumožňuje stanovení poloh hydridů v krystalech a k určení polohy hydridu blízko těžkého atomu je tak třeba použít jiný postup, například neutronovou difrakci nebo NMR.
- Klasický koncový hydrid: M—H
- Klasický můstkový: M—H—M
- Neklasický (divodík): M—H2
Spektroskopie
Hydridy pozdních přechodných kovů mají v protonové NMR signály M-H často mezi −5 a −25 pm; je známa i řada případů mimo toto rozmezí, ovšem vždy pod 0 ppm. Velké posuny jsou způsobeny vlivy excitovaných stavů a silným spinorbitálním párováním.[18] Krajním případem je 16elektronový komplex IrHCl2(PMe(t-Bu)2)2, s posunem −50,5 pm.
Signály často vykazují spin-spinová párování s jinými ligandy, jako jsou například fosfiny.[19]
Hydridy kovů mají v infračervených spektrech pásy kolem 2000 cm−1m odpovídající νM−H, jejich intenzity bývají různé.[4] Tyto signály lze identifikovat pomocí deuteriového značkování.
Historie
Špatně určitelné hydridy mědi byly objeveny v roce 1844 jako produkty reakcí solí mědi s kyselinou fosfornou. Následně bylo zjištěno, že plynný vodík může být absorbován směsmi Grignardových činidel a solí mědi.[20]
Prvním dobře popsatelným hydridokomplexem byl hydrid tetrakarbonylu železa, H2Fe(CO)4, vytvořený protonací aniontu karbonylu železa za nízké teploty. Dalším popsaným hydridovým komplexem se stal (C5H5)2ReH, zkoumaný NMR spektroskopií, která se tak ukázala být dobrým nástrojem pro výzkum hydridů kovů.[20] V roce 1957 popsali Joseph Chatt, Bernard L. Shaw a L. A. Duncanson trans-PtHCl(PEt3)2 jako první neorganokovový (tedy neobsahující vazbu kov–uhlík) hydrid. Tato látka byla na vzduchu stálá, což vyvrátilo představy o nestálosti kovových hydridů.[21]
Reference
V tomto článku byl použit překlad textu z článku Transition metal hydride na anglické Wikipedii.
- ↑ GREENWOOD, Norman; EARNSHAW, Alan. Chemistry of the Elements. 2. vyd. [s.l.]: Butterworth-Heinemann, 1997. ISBN 978-0-08-037941-8. (anglicky)
- ↑ S. C. Abrahams; A. P. Ginsberg; K. Knox. Transition Metal-Hydrogen Compounds. II. The Crystal and Molecular Structure of Potassium Rhenium Hydride, K2ReH9. Inorganic Chemistry. 1964, s. 558–567. DOI 10.1021/ic50014a026.
- ↑ R. B. King. Structure and bonding in homoleptic transition metal hydride anions. Coordination Chemistry Reviews. 2000, s. 813–829. DOI 10.1016/S0010-8545(00)00263-0.
- ↑ a b H. D. Kaesz; R. B. Saillant. Hydride complexes of the transition metals. Chemical Reviews. 1972, s. 231–281. DOI 10.1021/cr60277a003.
- ↑ Athanasios Zavras; George N. Khairallah; Timothy U. Connell; Jonathan M. White; Alison J. Edwards; Paul S. Donnelly; Richard A. J. O'Hair. Synthesis, Structure and Gas-Phase Reactivity of a Silver Hydride Complex [Ag3{(PPh2)2CH2}3(μ3-H)(μ3-Cl)]BF4. Angewandte Chemie. 2013-08-05, s. 8549–8552. ISSN 1521-3757. DOI 10.1002/ange.201302436.
- ↑ Elliot L. Bennett; Patrick J. Murphy; Silvia Imberti; Stewart F. Parker. Characterization of the Hydrides in Stryker's Reagent: [HCu{P(C6H5)3}]6. Inorganic Chemistry. 2014-03-17, s. 2963–2967. ISSN 0020-1669. DOI 10.1021/ic402736t. PMID 24571368.
- ↑ HARTWIG, John F. Organotransition Metal Chemistry: From Bonding to Catalysis. [s.l.]: University Science Books, 10. 2. 2010. 1172 s. Dostupné online. ISBN 1-891389-53-X. (anglicky)
- ↑ M. Tilset. Comprehensive Organometallic Chemistry III. [s.l.]: [s.n.], 2007. ISBN 9780080450476. Kapitola Organometallic Electrochemistry: Thermodynamics of Metal–Ligand Bonding, s. 279–305.
- ↑ M. Rakowski DuBois; D. L. DuBois. The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. zenodo.org. 2009, s. 62–72. Dostupné online. DOI 10.1039/b801197b.
- ↑ Danial D. M. Wayner; Vernon D. Parker. Bond Energies in Solution from Electrode Potentials and Thermochemical Cycles. A Simplified and General Approach. Accounts of Chemical Research. 1993, s. 287–294. DOI 10.1021/ar00029a010.
- ↑ M. Tilset "Organometallic Electrochemistry: Thermodynamics of Metal–Ligand Bonding" in Comprehensive Organometallic Chemistry III, Eds Crabtree, R. H.; Mingos, D. M. P. 2007 Elsevier ISBN 9780080445915
- ↑ J. R. Norton; K. W. Kramarz. Progress in Inorganic Chemistry. [s.l.]: [s.n.], 2007. ISBN 978-0-470-16643-7. Kapitola Slow Proton-Transfer Reactions in Organometallic and Bioinorganic Chemistry, s. 1–65.
- ↑ R. Bau; M. H. Drabnis. Structures of Transition Metal Hydrides Determined by Neutron Diffraction. Inorganica Chimica Acta. 1997, s. 27–50. DOI 10.1016/S0020-1693(97)89125-6.
- ↑ Pauline Chiu; Zhengning Li; Kelvin C. M. Fung. An expedient preparation of Stryker's reagent. Tetrahedron Letters. 2003, s. 455–457. Dostupné online [cit. 2009-04-17]. DOI 10.1016/S0040-4039(02)02609-6.
- ↑ S. Brayshaw; A. Harrison; J. McIndoe; F. Marken; P. Raithby; J. Warren; A. Weller. Sequential Reduction of High Hydride Count Octahedral Rhodium Clusters [Rh6(PR3)6H12][BArF4]2: Redox-Switchable Hydrogen Storage. Journal of the American Chemical Society. 2007, s. 1793–1804. DOI 10.1021/ja066940m. PMID 17284009.
- ↑ G. J. Kubas; R. R. Ryan; B. I. Swanson; P. J. Vergamini; H. J. Wasserman. Characterization of the first examples of isolable molecular hydrogen complexes, M(CO)3(PR3)2(H2) (M = molybdenum or tungsten; R = Cy or isopropyl). Evidence for a side-on bonded dihydrogen ligand. Journal of the American Chemical Society. 1984, s. 451–452. DOI 10.1021/ja00314a049.
- ↑ Gregory J. Kubas. Metal Dihydrogen and -Bond Complexes - Structure, Theory, and Reactivity. [s.l.]: Springer, 2001-08-31. ISBN 978-0-306-46465-2.
- ↑ P. Hrobarik; V. Hrobarikova; F. Meier; M. Repisky; S. Komorovsky; M. Kaupp. Relativistic Four-Component DFT Calculations of 1H NMR Chemical Shifts in Transition-Metal Hydride Complexes: Unusual High-Field Shifts Beyond the Buckingham–Stephens Model. Journal of Physical Chemistry A. 2011, s. 5654–5659. DOI 10.1021/jp202327z. PMID 21591659. Bibcode 2011JPCA..115.5654H.
- ↑ J. W. Akitt in "Multinuclear NMR" Joan Mason (Editor), 1987, Plenum Press ISBN 0-306-42153-4
- ↑ a b Joseph Chatt. Hydride Complexes. Science. 1968, s. 723–729. DOI 10.1126/science.160.3829.723. PMID 17784306. Bibcode 1968Sci...160..723C.
- ↑ J. Chatt; L. A. Duncanson; B. L. Shaw. A Volatile Chlorohydride of Platinum. Proceedings of the Chemical Society. 1957, s. 329–368. DOI 10.1039/PS9570000329.
Portály: Chemie










