Reakcja Stettera
Reakcja Stettera – reakcja chemiczna tworzenia wiązań węgiel–węgiel przez w wyniku 1,4-addycji aldehydów z α,β-nienasyconymi ketonami (w podstawowej wersji), z wykorzystaniem katalizatora nukleofilowego[1]. Podczas gdy podobna reakcja 1,2-addycji, kondensacja benzoinowa(inne języki), była znana już w latach 30. XIX w., pierwszy przykład reakcji nazwanej później reakcją Stettera, został opisany dopiero w 1973 r. przez Hermanna Stettera(inne języki)[2]. Reakcja ta pozwala na utworzenie związków 1,4-dikarbonylowych z aldehydów oraz akceptorów Michaela. W przeciwieństwie do 1,3-dikarbonyli, które można otrzymać w reakcji kondensacji Claisena, i 1,5-dikarbonyli, które można otrzymać w reakcji Michaela, związki 1,4-dikarbonylowe są trudniejsze do otrzymania, ale są cennymi półproduktami m.in. w otrzymywaniu furanów, piroli i tiofenów w reakcji Paala-Knorra. Typowymi katalizatorami tej reakcji są sole tiazolowe i anion cyjankowy, ale w przypadkach asymetrycznej reakcji Stettera używane są sole triazolowe. Reakcja Stettera jest przykładem reakcji wprowadzającej umpolung(inne języki), tj. odwrócenie polarności, ponieważ zazwyczaj elektrofilofilowy węgiel karbonylowy aldehydu zachowuje się w tym przypadku jako nukleofil.

Mechanizm
Ponieważ reakcja Stettera zachodzi z odwróceniem polarności, aldehyd musi zostać przejściowo zamieniony z elektrofila w nukleofil[3]. Dzieje się to przez przyłączenie węgla karbonylowego do katalizatora – cyjanku lub N-heterocyklicznego karbenu, utworzonego zazwyczaj in situ z soli tiazolowej lub triazolowej i odpowiedniej zasady[1]. W obu przypadkach mechanizm jest podobny: nukleofilowy atom węgla z cyjanku lub N-heterocyklicznego karbenu przyłącza się do atomu węgla karbonylowego, co powoduje powstanie produktu pośredniego – cyjanohydryny lub tzw. produktu Breslowa – w którym były węgiel karbonylowy może być deprotonowany (cyjanohydryna) lub tworzy enolan (produkt Breslowa), w którym były węgiel karbonylowy również jest nukleofilowy. Taki związek został zaproponowany przez Ronalda Breslowa w 1958 roku jako wspólny produkt pośredni dla wszystkich reakcji katalizowanych solami tiazolowymi, w tym tiaminą, zarówno in vitro, jak i in vivo[4].

Tak utworzony synton „nukleofilowego aldehydu” może reagować na dwa sposoby. Jednym z nich jest kondensacja z kolejnym aldehydem i wytworzeniem produktu kondensacji benzoinowej, jednak ponieważ reakcja ta jest odwracalna w tych warunkach, nie przeszkadza to w zajściu wolniejszej reakcji Stettera. Wykorzystując tę odwracalność, reakcję Stettera można przeprowadzić z benzoinami zamiast z aldehydami[1]. Właściwa reakcja Stettera zachodzi przez addycję równoważnika nukleofilowego aldehydu do akceptora Michaela. Ten etap jest nieodwracalny i w następnej kolejności powstaje 1,4-dikarbonyl z odtworzeniem katalizatora.

Zakres stosowalności reakcji
Reakcja Stettera dostarcza trudne do uzyskania innymi drogami związki 1,4-dikarbonylowe i ich pochodne. Typowe warunki pozwalają na użycie wielu różnych substratów[1]. Jako źródła anionu acylowego mogą być użyte aldehydy aromatyczne i heteroaromatyczne oraz odpowiednie benzoiny, zarówno z solami tiazolowymi, jak i cyjankiem jako katalizatorami. Aldehydy alkilowe mogą być użyte tylko z solami tiazolowymi, ponieważ cyjanek jest w tym przypadku zbyt silną zasadą, co powoduje powstawanie produktów kondensacji aldolowej. Jako akceptory Michaela mogą zostać użyte Estry α,β-nienasycone, enony, enale, nienasycone nitryle i nitrozwiązki. Co do zasady, w asymetrycznej wersji reakcji Stettera używa się węższego zakresu substratów. Natomiast w wewnątrzcząsteczkowych asymetrycznych reakcjach Stettera można używać wiele typowych akceptorów Michaela i aldehydów w niemal dowolnych kombinacjach[5]. W takiej reakcji można zastosować zarówno aldehydy aromatyczne, heteroaromatyczne, jak i alkilowe z akceptorem Michaela w tej samej cząsteczce. Odpowiednie akceptory Michaela obejmują α,β-nienasycone estry, ketony, tioestry, nitryle, amidy Weinreba oraz alkilidenomaloniany. Nitroalkeny i enale ulegają reakcjom konkurencyjnym i nie pozwalają na uzyskanie produktu reakcji Stettera[6]. Zarówno produkty z pierścieniem pięcioczłonowym[7][8], jak i sześcioczłonowym[9][10] można uzyskać z wysokimi wydajnościami i nadmiarami enancjomerycznymi; produkty z pierścieniem siedmioczłonowym nie zostały do tej pory uzyskane w ten sposób[5]. W porównaniu do reakcji wewnątrzcząsteczkowych, asymetryczne międzycząsteczkowe reakcje Stettera są mniej zbadane i opracowane zostały dla ściśle dopasowanych par akceptora Michaela i aldehydu. W jednym przypadku literaturowym użyto aldehydu alifatycznego i nitroalkenu[11].

Modyfikacje
Od czasu odkrycia reakcji Stettera opisanych zostało wiele jej modyfikacji. W 2001 roku Jerry A. Murry, Doug E. Frantz i in. opublikowali przykład reakcji aza-Stettera między aldehydami aromatycznymi a N-acyloiminami dającej α-amidoketony[12]. N-Acyloiminy zostały w tym przypadku utworzone in situ z adduktów sulfinowych, które ulegają eliminacji do imin w warunkach zasadowych. Reakcja ta pozwala na uzyskanie dobrych wydajności (75–90%); odpowiednie benzoiny nie są w tym przypadku dobrymi substratami[12], w przeciwieństwie do bardziej typowych reakcji Stettera[1]. Autorzy wnioskują z tego, że w tym przypadku reakcja Stettera jest pod kontrolą kinetyczną, w przeciwieństwie do kontroli termodynamicznej w normalnych warunkach[12]:

Inną modyfikacją jest użycie α-ketokwasów jako prekursorów anionu acylowego. W 2005 roku została opisana reakcja wykorzystująca pirogronian sodu zamiast aldehydu; reakcji utworzenia produktu Breslowa towarzyszy dekarboksylacja[13]. W podobny sposób można użyć 1,2-diketonów: w 2011 roku ukazała się praca wykorzystująca 2,3-butadion jako źródło anionu acylowego. W warunkach opracowanych przez autorów 2,3-butanedion rozszczepia się na octan etylu oraz produkt Breslowa uczestniczący w reakcji Stettera[14]:

W tej samej pracy zademonstrowano użycie cyklicznego 1,2-diketonu jako źródła anionu acylowego w reakcji Stettera, jednocześnie dostarczającego terminalnego estru etylowego w produkcie. Reakcja zachodzi jak z 2,3-butadionem, oprócz tego, że ester pozostaje w cząsteczce i nie jest produktem ubocznym[14]:
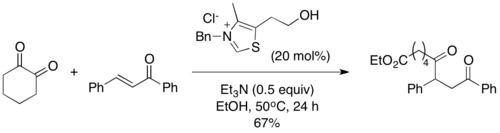
Uzyskano w ten sposób tylko estry etylowe; użycie tert-butanolu skończyło się niepowodzeniem[14].
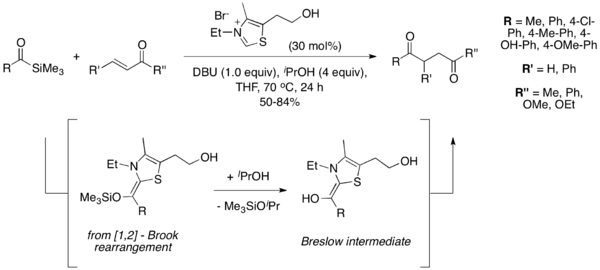
W roku 2004 Karl A. Scheidt i wsp. jak źródła anionu acylowego użył acylosilanów w reakcji Stettera. Modyfikacja ta nosi nazwę „reakcja sila-Stettera”[15]. W tych warunkach katalizator tiazolowy powoduje zajście przegrupowania [1,2]-Brooka(inne języki), po którym za pomocą izopropanolu przeprowadza się desilylację, co pozwala na uzyskanie produktu Breslowa. Reakcja nie zachodzi bez dodatku alkoholu, co wskazuje na konieczność zajścia desilylacji w celu uzyskania produktu. Acylosilany są mniej elektrofilowe niż odpowiednie aldehydy, co pozwala na uniknięcie powstawania produktów reakcji benzoinowej[16].
Asymetryczna reakcja Stettera
Pierwszy wariant asymetryczny reakcji Stettera został opublikowany w 1996 przez Endersa wykorzystując chiralny katalizator triazolowy (związek 1 na ilustracji poniżej)[17]. W późniejszych publikacjach zostały opisane kolejne katalizatory asymetrycznej reakcji Stettera, takie jak 2[18], 3[19], i 4[20]:

Szczególnie wartościowy okazał się katalizator Rovisa 2. W dalszej kolejności autorzy modyfikowali reszty arylowe zachowując szkielet aminoindanolu, co pozwoliło im na przeprowadzenie enancjoselektywnej wewnątrzcząsteczkowej reakcji Stettera tworzącej czwartorzędowe centrum stereogeniczne z aldehydami aromatycznymi jako substratami[21]. Dalsze prace pozwoliły również na użycie aldehydów alkilowych[22]. Dokładniejsze badania pozwoliły na ustalenie, że geometria substratu decyduje o diasteroselektywności, natomiast od użytego katalizatora zależy enancjoselektywność reakcji. W pierwszej kolejności następuje ustalenie konfiguracji węgla β z akceptora Michaela (przez addycję nukleofilowego aldehydu), a następnie węgla α (przez diastereoselektywne protonowanie powstałego anionu)[23]:

Nieodłączne trudności z kontrolą enancjoselektywności w międzycząsteczkowej reakcji Stettera sprawiają, że opracowywanie warunków tego wariantu reakcji pozostaje dużym wyzwaniem. Podczas gdy pewne ograniczone nadmiary enancjomeryczne zostały uzyskane przez Endersa w latach 90. dla reakcji n-butanalu z chalkonem[24], syntetycznie użyteczne warunki reakcji nie zostały uzyskane aż do 2008 roku. Grupa Dietera Endersa opisała wówczas użycie katalizatora triazolowego w reakcji aldehydów aromatycznych z chalkonami, uzyskując umiarkowane wydajności[25]. W tym samym czasie ukazała się publikacja grupy Tomislava Rovisa, opisująca sprzęganie glioksamidu z alkilidenomalonianami w obecności katalizatora triazolowego, z wysokimi wydajnościami[26]:

Rovis i wsp. zbadali następnie asymetryczną międzycząsteczkową reakcję Stettera aldehydów heterocyklicznych i nitroalkenów[27]. Podczas optymalizacji tej reakcji okazało się, że najwyższą enancjoselektywność zapewnia katalizator z podstawnikiem fluorowym. Autorzy sugerują, że pomaga on usztywnić konformację katalizatora w taki sposób, który zwiększa enancjoselektywność. Badania obliczeniowe wskazują, że większy nadmiary enancjomeryczy jest efektem oddziaływania między częściowym ładunkiem dodatnim wiązania C−F i powstającym podczas reakcji ładunkiem ujemnym na nitroalkenie[28].

Innym przykładem asymetrycznej reakcji Stettera jest enancjoselektywna synteza α-aminokwasów z wykorzystaniem N-acylamidoakrylanu jako akceptora Michaela[11]:

Zastosowania
Reakcja Stettera jest użytecznym narzędziem w syntezie organicznej. Produkty tej reakcji, związki 1,4-dikarbonylowe, mogą być wprowadzane do bardziej złożonych związków. W jednym z zastosowań Trost użył reakcji Stettera w syntezie totalnej produktu naturalnego, racemicznego kwasu hirsutowego C. Wewnątrzcząsteczkowe sprzęganie aldehydu alifatycznego z α,β-nienasyconym estrem pozwoliło na otrzymanie tricyklicznego 1,4-dikarbonylu z wydajnością 67%. W siedmiu kolejnych etapach przekształcono ten związek w kwas hirsutowy C[29]:

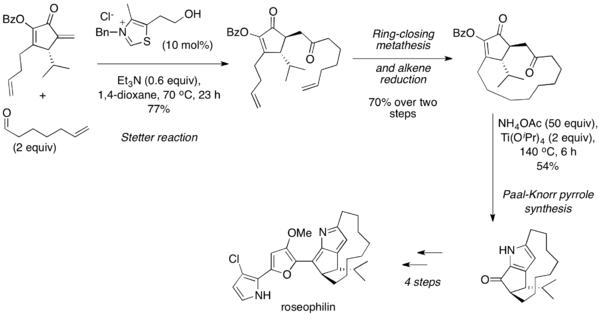
Reakcja Stettera jest często wykorzystywana do syntezy substratów do syntezy Paala-Knorra, takich jak furany, tiofeny i pirole. W 2001 roku Marcus A. Tius zaprezentował asymetryczną syntezę totalną roseofiliny, wykorzystującą wewnątrzcząsteczkową reakcję Stettera[30]. Dalsza reakcja metatezy olefin i redukcja alkenu pozwoliła na uzyskanie 1,4-dikarbonylu będącego substratem reakcji Paala-Knorra prowadzącej do pirolu. Kolejne przekształcenia pozwoliły na uzyskanie roseofiliny:
W 2004 roku została zaprezentowana sekwencja reakcji one-pot (tj. bez izolowania produktów pośrednich): sprzęgania z kompleksami palladu – izomeryzacja – reakcja Stettera – reakcja Paala-Knorra[31]. W pierwszej kolejności zachodzi sprzęganie halogenków arylowych z alkoholami propargilowymi, co daje α,β-nienasycone ketony. Te enony ulegają reakcji Stettera z aldehydami, natomiast w odpowiednich warunkach produkt 1,4-dikarbonylowy ulega reakcji Paala-Knorra do furanu lub pirolu.

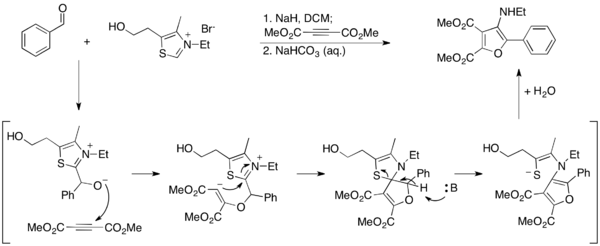
Cheng Ma i wsp. opracowali alternatywną metodę syntezy furanów z wykorzystaniem reakcji Stettera[32]. W ich podejściu 3-aminofurany powstają w warunkach reakcji Stettera między aldehydami aromatycznymi i dimetyloacetylenodikarboksylanem, gdzie karben tiazolowy jest hydrolizowany równocześnie z aromatyzacją produktu do furanu. Ponieważ pierścień tiazolowy ulega w tym przypadku rozkładowi, reakcja nie jest katalityczna i sól tiazolowa musi być użyta w ilościach stechiometrycznych:
Dalsze prace w tym kierunku pozwoliły na uzyskanie 2-aminofuranów przez reakcję wykorzystującą nitryle[33]. W tym przypadku sól tiazolowa jest zastosowana w ilościach katalitycznych:

Zobacz też
Przypisy
- ↑ a b c d e HermannH. Stetter HermannH., Catalyzed Addition of Aldehydes to Activated Double Bonds?A New Synthetic Approach, „Angewandte Chemie International Edition in English”, 15 (11), 1976, s. 639–647, DOI: 10.1002/anie.197606391 (ang.).
- ↑ HermannH. Stetter HermannH., ManfredM. Schreckenberg ManfredM., A New Method for Addition of Aldehydes to Activated Double Bonds, „Angewandte Chemie International Edition in English”, 12 (1), 1973, s. 81–81, DOI: 10.1002/anie.197300811 (ang.).
- ↑ J. DonaldJ.D. Albright J. DonaldJ.D., Reactions of acyl anion equivalents derived from cyanohydrins, protected cyanohydrins and α-dialkylaminonitriles, „Tetrahedron”, 39 (20), 1983, s. 3207–3233, DOI: 10.1016/S0040-4020(01)91568-6 (ang.).
- ↑ RonaldR. Breslow RonaldR., On the Mechanism of Thiamine Action. IV.1 Evidence from Studies on Model Systems, „Journal of the American Chemical Society”, 80 (14), 1958, s. 3719–3726, DOI: 10.1021/ja01547a064 (ang.).
- ↑ a b Javier Read deJ.R. Alaniz Javier Read deJ.R. i inni, Scope of the Asymmetric Intramolecular Stetter Reaction Catalyzed by Chiral Nucleophilic Triazolinylidene Carbenes, „Journal of Organic Chemistry”, 73 (6), 2008, s. 2033–2040, DOI: 10.1021/jo702313f, PMID: 18302407, PMCID: PMC4222522 (ang.).
- ↑ Mark S.M.S. Kerr Mark S.M.S., TomislavT. Rovis TomislavT., Effect of the Michael Acceptor in the Asymmetric Intramolecular Stetter Reaction, „Synlett”, 2003 (12), 2003, s. 1934–1936, DOI: 10.1055/s-2003-41458 (ang.).
- ↑ Mark S.M.S. Kerr Mark S.M.S., TomislavT. Rovis TomislavT., Enantioselective Synthesis of Quaternary Stereocenters via a Catalytic Asymmetric Stetter Reaction, „Journal of the American Chemical Society”, 126 (29), 2004, s. 8876–8877, DOI: 10.1021/ja047644h .
- ↑ QinQ. Liu QinQ., TomislavT. Rovis TomislavT., Enantioselective Synthesis of Hydrobenzofuranones Using an Asymmetric Desymmetrizing Intramolecular Stetter Reaction of Cyclohexadienones, „Organic Process Research & Development”, 11 (3), 2007, s. 598–604, DOI: 10.1021/op600278f, PMID: 19603085, PMCID: PMC2709411 (ang.).
- ↑ Mark S.M.S. Kerr Mark S.M.S., Javier Read deJ.R. Alaniz Javier Read deJ.R., TomislavT. Rovis TomislavT., A Highly Enantioselective Catalytic Intramolecular Stetter Reaction, „Journal of the American Chemical Society”, 124 (35), 2002, s. 10298–10299, DOI: 10.1021/ja027411v (ang.).
- ↑ TaijuT. Nakamura TaijuT. i inni, A Facile Synthesis of Chroman-4-ones and 2,3-Dihydroquinolin-4-ones with Quaternary Carbon Using Intramolecular Stetter Reaction Catalyzed by Thiazolium Salt, „Synlett”, 2005 (01), 2005, s. 155–157, DOI: 10.1055/s-2004-835666 (ang.).
- ↑ a b ThierryT. Jousseaume ThierryT., Nathalie E.N.E. Wurz Nathalie E.N.E., FrankF. Glorius FrankF., Highly Enantioselective Synthesis of α-Amino Acid Derivatives by an NHC-Catalyzed Intermolecular Stetter Reaction, „Angewandte Chemie International Edition”, 50 (6), 2011, s. 1410–1414, DOI: 10.1002/anie.201006548 (ang.).
- ↑ a b c Jerry A.J.A. Murry Jerry A.J.A. i inni, Synthesis of α-Amido Ketones via Organic Catalysis: Thiazolium-Catalyzed Cross-Coupling of Aldehydes with Acylimines, „Journal of the American Chemical Society”, 123 (39), 2001, s. 9696–9697, DOI: 10.1021/ja0165943, PMID: 11572700 (ang.).
- ↑ Michael C.M.C. Myers Michael C.M.C. i inni, Catalytic Conjugate Additions of Carbonyl Anions under Neutral Aqueous Conditions, „Journal of the American Chemical Society”, 127 (42), 2005, s. 14675–14680, DOI: 10.1021/ja0520161, PMID: 16231921 (ang.).
- ↑ a b c OlgaO. Bortolini OlgaO. i inni, Thiazolium-catalyzed intermolecular Stetter reaction of linear and cyclic alkyl α-diketones, „Organic & Biomolecular Chemistry”, 9 (24), 2011, s. 8437, DOI: 10.1039/c1ob06480k (ang.).
- ↑ Anita E.A.E. Mattson Anita E.A.E., Ashwin R.A.R. Bharadwaj Ashwin R.A.R., Karl A.K.A. Scheidt Karl A.K.A., The Thiazolium-Catalyzed Sila-Stetter Reaction: Conjugate Addition of Acylsilanes to Unsaturated Esters and Ketones, „Journal of the American Chemical Society”, 126 (8), 2004, s. 2314–2315, DOI: 10.1021/ja0318380, PMID: 14982429 (ang.).
- ↑ Anita E.A.E. Mattson Anita E.A.E. i inni, Thiazolium-Catalyzed Additions of Acylsilanes: A General Strategy for Acyl Anion Addition Reactions, „Journal of Organic Chemistry”, 71 (15), 2006, s. 5715–5724, DOI: 10.1021/jo060699c, PMID: 16839153 (ang.).
- ↑ DieterD. Enders DieterD. i inni, The First Asymmetric IntramolecularStetter Reaction. Preliminary Communication, „Helvetica Chimica Acta”, 79 (7), 1996, s. 1899–1902, DOI: 10.1002/hlca.19960790712 (ang.).
- ↑ Mark S.M.S. Kerr Mark S.M.S., Javier Read deJ.R. Alaniz Javier Read deJ.R., TomislavT. Rovis TomislavT., A Highly Enantioselective Catalytic Intramolecular Stetter Reaction, „Journal of the American Chemical Society”, 124 (35), 2002, s. 10298–10299, DOI: 10.1021/ja027411v, PMID: 12197730 (ang.).
- ↑ JensJ. Pesch JensJ., KlausK. Harms KlausK., ThorstenT. Bach ThorstenT., Preparation of Axially ChiralN,N′-Diarylimidazolium andN-Arylthiazolium Salts and Evaluation of Their Catalytic Potential in the Benzoin and in the Intramolecular Stetter Reactions, „European Journal of Organic Chemistry”, 2004 (9), 2004, s. 2025–2035, DOI: 10.1002/ejoc.200300762 (ang.).
- ↑ Steven M.S.M. Mennen Steven M.S.M. i inni, A peptide-catalyzed asymmetric Stetter reaction, „Chemical Communications” (2), 2005, s. 195–197, DOI: 10.1039/B414574G, PMID: 15724183 (ang.).
- ↑ Mark S.M.S. Kerr Mark S.M.S., TomislavT. Rovis TomislavT., Enantioselective Synthesis of Quaternary Stereocenters via a Catalytic Asymmetric Stetter Reaction, „Journal of the American Chemical Society”, 126 (29), 2004, s. 8876–8877, DOI: 10.1021/ja047644h, PMID: 15264801 (ang.).
- ↑ Jennifer L.J.L. Moore Jennifer L.J.L., Mark S.M.S. Kerr Mark S.M.S., TomislavT. Rovis TomislavT., Enantioselective formation of quaternary stereocenters using the catalytic intramolecular Stetter reaction, „Tetrahedron”, 62 (49), 2006, s. 11477–11482, DOI: 10.1016/j.tet.2006.06.042 (ang.).
- ↑ Javier Read deJ.R. Alaniz Javier Read deJ.R., TomislavT. Rovis TomislavT., A Highly Enantio- and Diastereoselective Catalytic Intramolecular Stetter Reaction, „Journal of the American Chemical Society”, 127 (17), 2005, s. 6284–6289, DOI: 10.1021/ja0425132, PMID: 15853335 (ang.).
- ↑ D.D. Enders D.D., Enzymemimetic C−C and C−N Bond Formations, [w:] EckhardE. Ottow, KlausK. Schöllkopf, Bernd-GünterB.G. Schulz (red.), Stereoselective Synthesis. Lectures honouring Prof. Dr. Dr. h.c. Rudolf Wiechert, Berlin, Heidelberg: Springer Berlin Heidelberg, 1993, s. 63–90, DOI: 10.1007/978-3-642-78496-5_4, ISBN 978-3-642-78496-5, OCLC 840299291 (ang.).
- ↑ DieterD. Enders DieterD., JianweiJ. Han JianweiJ., AlexanderA. Henseler AlexanderA., Asymmetric intermolecular Stetter reactions catalyzed by a novel triazolium derived N-heterocyclic carbene, „Chemical Communications” (34), 2008, s. 3989, DOI: 10.1039/b809913h, PMID: 18758602 (ang.).
- ↑ QinQ. Liu QinQ., StéphaneS. Perreault StéphaneS., TomislavT. Rovis TomislavT., Catalytic Asymmetric Intermolecular Stetter Reaction of Glyoxamides with Alkylidenemalonates, „Journal of the American Chemical Society”, 130 (43), 2008, s. 14066–14067, DOI: 10.1021/ja805680z, PMID: 18834123, PMCID: PMC2684863 (ang.).
- ↑ Daniel A.D.A. DiRocco Daniel A.D.A. i inni, Catalytic Asymmetric Intermolecular Stetter Reaction of Heterocyclic Aldehydes with Nitroalkenes: Backbone Fluorination Improves Selectivity, „Journal of the American Chemical Society”, 131 (31), 2009, s. 10872–10874, DOI: 10.1021/ja904375q, PMID: 19722669, PMCID: PMC2747345 (ang.).
- ↑ Joann M.J.M. Um Joann M.J.M. i inni, Quantum Mechanical Investigation of the Effect of Catalyst Fluorination in the Intermolecular Asymmetric Stetter Reaction, „Journal of the American Chemical Society”, 133 (29), 2011, s. 11249–11254, DOI: 10.1021/ja202444g, PMID: 21675770, PMCID: PMC3143204 (ang.).
- ↑ Barry M.B.M. Trost Barry M.B.M., Charles D.Ch.D. Shuey Charles D.Ch.D., FrankF. DiNinno FrankF., A stereocontrolled total synthesis of (±)-hirsutic acid C, „Journal of the American Chemical Society”, 101 (5), 1979, s. 1284–1285, DOI: 10.1021/ja00499a043 (ang.).
- ↑ Paul E.P.E. Harrington Paul E.P.E., Marcus A.M.A. Tius Marcus A.M.A., Synthesis and Absolute Stereochemistry of Roseophilin, „Journal of the American Chemical Society”, 123 (35), 2001, s. 8509–8514, DOI: 10.1021/ja011242h, PMID: 11525658 (ang.).
- ↑ Roland U.R.U. Braun Roland U.R.U., Thomas J.T.J. Müller Thomas J.T.J., Coupling-Isomerization-Stetter and Coupling-Isomerization-Stetter-Paal-Knorr Sequences – A Multicomponent Approach to Furans and Pyrroles, „Synthesis”, 2004 (14), 2004, s. 2391–2406, DOI: 10.1055/s-2004-831192 (niem.).
- ↑ ChengCh. Ma ChengCh., YeweiY. Yang YeweiY., Thiazolium-Mediated Multicomponent Reactions: A Facile Synthesis of 3-Aminofuran Derivatives, „Organic Letters”, 7 (7), 2005, s. 1343–1345, DOI: 10.1021/ol0501368, PMID: 15787502 (ang.).
- ↑ PengP. Liu PengP. i inni, An Efficient Synthesis of 2-Aminofuran-3-carbonitriles via Cascade Stetter-γ-Ketonitrile Cyclization Reaction Catalyzed by N-Heterocyclic Carbene, „Synlett”, 2011 (08), 2011, s. 1133–1136, DOI: 10.1055/s-0030-1259945 (ang.).












